The Therapeutically Applicable Research to Generate Effective Treatments (TARGET) program applies a comprehensive genomic approach to determine molecular changes that drive childhood cancers. The goal of the program is to use data to guide the development of effective, less toxic therapies. TARGET is organized into a collaborative network of disease-specific project teams.
TARGET AML Overview¶
The TARGET Acute Myeloid Leukemia (AML) projects elucidate comprehensive molecular characterization to determine the genetic changes that drive the initiation and progression of high-risk or hard-to-treat childhood cancers. Acute myeloid leukemia (AML) is a cancer that originates in the bone marrow from immature white blood cells known as myeloblasts. About 25% of all children with leukemia have AML.
TARGET investigators are analyzing tumors from pediatric patients, many who have relapsed, to identify biomarkers that correlate with poor clinical outcome and/or new therapeutic approaches to treat childhood AML. The tissues used in this study were collected from patients enrolled in Children's Oncology Group (COG) biology studies and clinical trials.
The AML project team members (like other TARGET researchers) are generating data in two phases: Discovery and Validation.
Research Methodology¶
TARGET researchers utilized various sequencing and array-based methods to examine the genomes, transcriptomes, and for some diseases epigenomes of select childhood cancers. This “multi-omic” approach generated comprehensive profiles of molecular alterations for each cancer type. Alterations are changes in DNA or RNA, such as rearrangements in chromosome structure or variations in gene expression, respectively. Through computational analyses and assays to validate biological function, TARGET researchers predict which alterations disrupt the function of a gene or pathway and promote cancer growth, progression, and/or survival. Researchers identify candidate therapeutic targets and/or prognostic markers from the cancer-associated alterations.
To learn about individual sequencing and array-based methods used in TARGET research, visit Methods below.
Discovery¶
TARGET project teams have characterized “discovery” cohorts of patient cases to identify molecular alterations of the transcriptome, genome, and epigenome in various pediatric cancer subtypes. Each project team independently selected their patient cohorts based upon characteristics of the disease or cancer subtype. A comprehensive genomic profile of each patient case was generated using nucleic acids from tumor tissue taken at the time of diagnosis and case-matched “normal” tissue. Whenever available, case-matched tissues from relapsed or treatment-resistant tumors were also characterized by the same methods.* Robust clinical data were obtained for each case studied in TARGET. All tissues used meet strict scientific, technical, and ethical requirements.
Verification¶
TARGET project teams employed multiple technologies to confirm the presence of mutations found in tumor tissue. The use of diverse sequencing approaches (e.g. mRNA-seq, whole exome or whole genome sequencing) to analyze discovery samples provided confirmation of somatic mutation(s) observed in a patient case and affirmed the quality of each data type generated. Individual project teams also analyzed a subset of their discovery cases using the method(s) employed for validation*.
Validation¶
TARGET project teams used a separate sequencing method applied to an independent cohort of patients to validate at the gene level most candidate mutations found in the TARGET discovery data. Validation confirmed the population prevalence of mutations. Within each disease, characterizing a separate cohort more broadly representative of the disease population further allowed the project teams to estimate the frequency of somatic variants in a given cancer subtype.
Experimental Methodology¶
Each project team employed a large subset of the methods below. Visit the TARGET Project Experimental Methods page for detailed protocols and to learn which methods apply for each individual project.
Genome-scale Characterization¶
- Gene expression profiling: determines patterns of all genes transcribed
- Copy number analysis: determines structural changes of chromosomes, such as copy number alterations (chromosome region gains and losses) including the loss of heterozygosity and translocations
- DNA methylation status: determines patterns of DNA (cytosine) methylation on chromosomes
- miRNA profiling: determines expression patterns of small regulatory molecules called microRNAs (miRNAs)
Sequencing¶
Sanger Method¶
- Targeted Sequencing: DNA sequencing of specific genes or areas of the genome
- Kinome Sequencing: DNA sequencing of genes encoding kinases
Next-generation Method¶
- Whole Genome Sequencing: provides the DNA sequence of the genome
- Whole Exome Sequencing: provides DNA sequences of exons
- Transcriptome Sequencing (RNA-seq): provides sequences from transcribed RNAs
- mRNA Sequencing (mRNA-seq): generates sequences of messenger RNAs (mRNAs)
- miRNA Sequencing (miRNA-seq): generates sequences of small RNA molecules, such as microRNAs(miRNAs)
- Bisulfite sequencing: identifies pattern of methylation of individual cytosines in DNA within a defined area of the genome (i.e. epigenome)
- ChIP sequencing: identifies protein interactions with DNA of the genome
Detailed Methodology¶
TARGET Dataset¶
Overview¶
The TARGET Data Matrix contains a summary of all the datasets associated with each of the TARGET project, including the TARGET AML project.
Data Types¶
The TARGET program outputs consist of two overall types of datasets: Patient Clinical Data and Patient Sample Data
Patient Clinical Data¶
Patient clinical data, which include patient demographics, can be found: - target-data.nci.nih.gov/Public/AML/clinical/harmonized/ [Google Drive Mirror]
Patient Sample Data¶
The TARGET AML project was broken into two phases, Discovery and Validation, which constitute two distinct datasets. Summaries of the data outputs of each phase are documented in the below samples matrices: - TARGET_AML_SampleMatrix_Discovery_20190807 - TARGET_AML_SampleMatrix_Validation_20180914
Patient Clinical Data¶
- TARGET_AML_CDE_20181213: Contains column metadata
- TARGET_AML_ClinicalData_AML1031_20190802: Patient clinical information
- ...
Patient Sample Data¶
Patient sample data is located in three locations: - TARGET Public Website - Globus Connect - Genomic Data Commons Data (GDC) Portal
Overall Dataset Information¶
TARGET Dataset Analysis¶
Directories¶
Naming Convention¶
In general, data stored at the DCC is organized within directories that correspond to the types of analysis, levels of data, and the names of centers that submitted a particular type of data when there are multiple data-submitting centers.
- Raw or low-level data files (level 1)
- Normalized and integrated data (levels 2 and 3)
- Summarized findings (level 4)
Structure¶
├── WGS (Whole Genome Sequence)
│ ├── CGI (Complete Genomics Inc)
│ │ ├── OptionAnalysisPipeline2
│ │ ├── PilotAnalysisPipeline2
│ │ ├── READMEs
│ │ └── RequestedReports
│ └── L3 (Level 3 Analysis)
│ ├── mutation
│ │ ├── BCCA (British Columbia Cancer Agency)
│ │ ├── CGI (Complete Genomics Inc)
│ │ │ ├── Analysis
│ │ │ ├── FullMafsVcfs
│ │ │ └── SomaticFilteredMafs
│ │ └── TAWG-StJude
│ │ └── CandidateSomatic
│ └── structural
│ ├── BCCA (British Columbia Cancer Agency)
│ ├── CGI (Complete Genomics Inc)
│ └── StJude
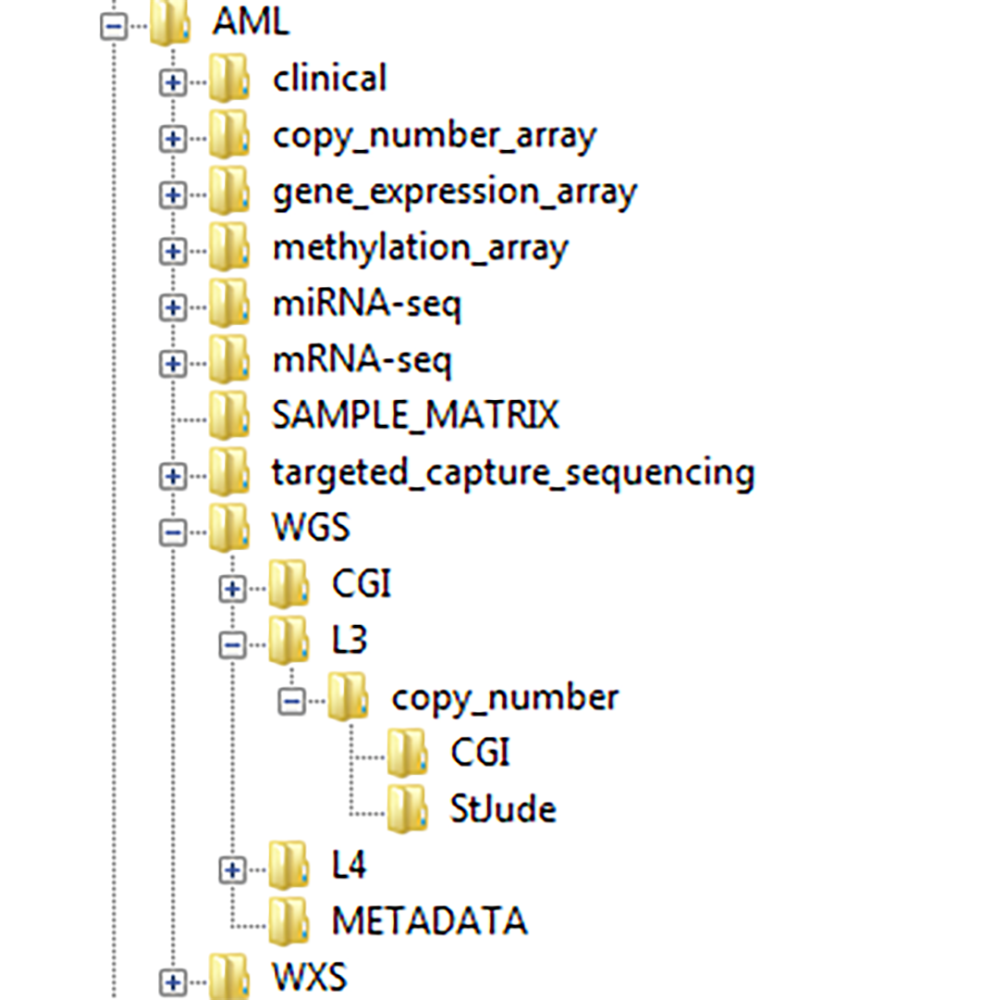 Figure 1: Example of a TARGET Project's download directory structure.
Figure 1: Example of a TARGET Project's download directory structure.
References¶
Files¶
Naming Convention¶
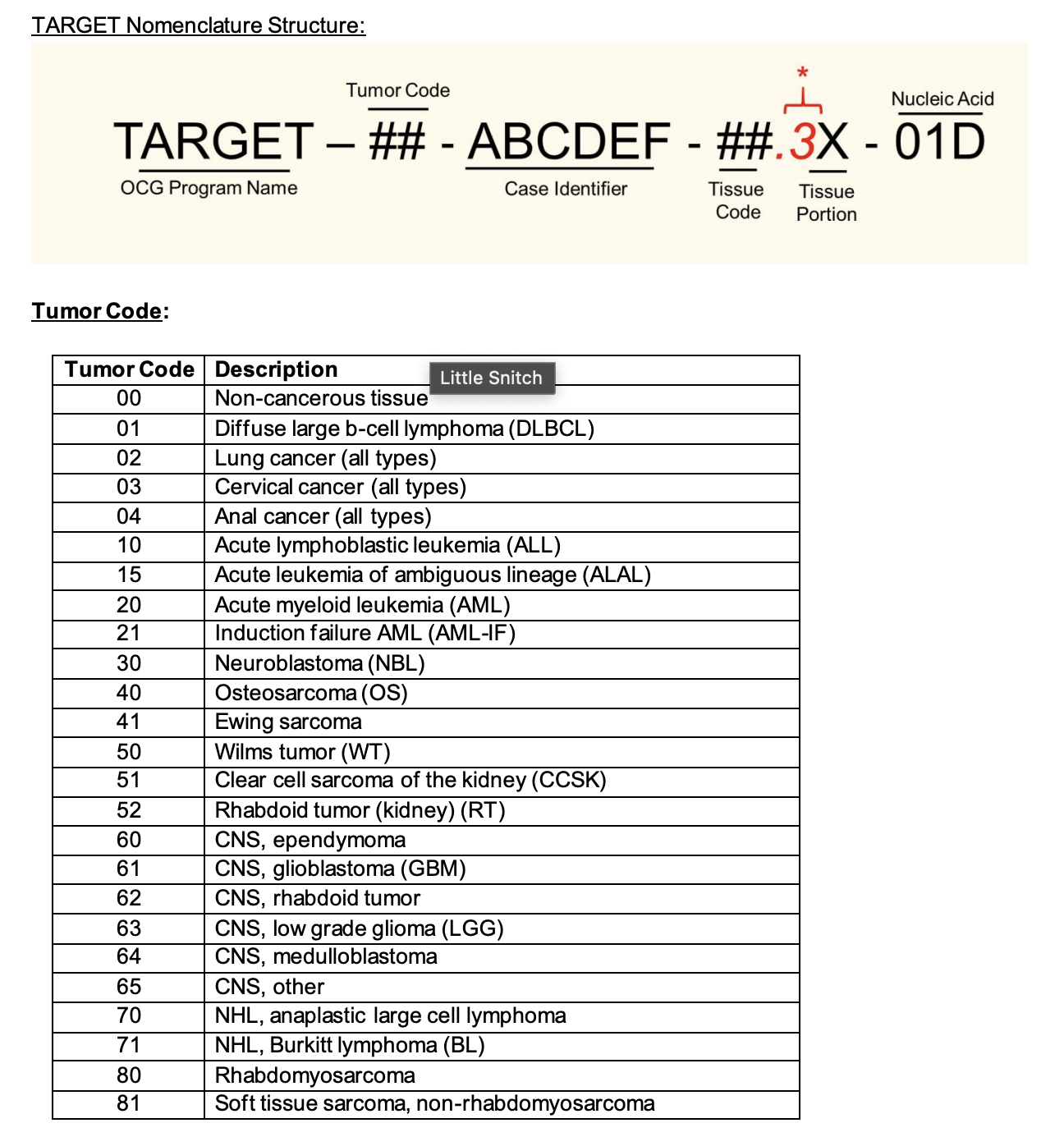
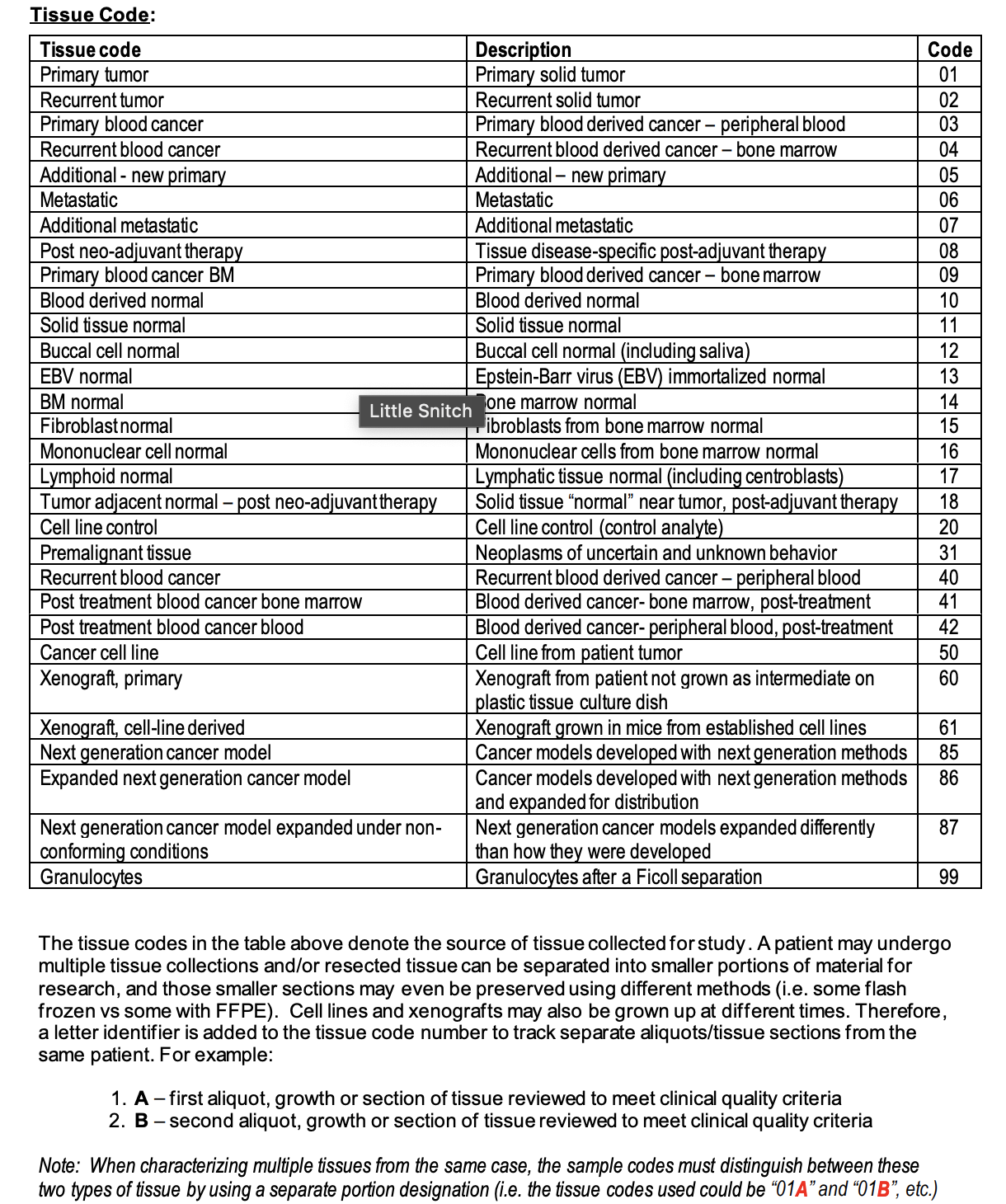

File Format Information¶
WGS Files¶
The Whole Genome Sequence (WGS) data was created by Complete Genomics Incorporated (CGI) and is delivered as a package, which is detailed here.
The packages are accessible on Globus, in the /AML/WGS/CGI/ directory.
Alternatively,
VCF Files¶
TSV Files¶
- https://stjudecloud.github.io/docs/guides/proteinpaint/file-formats/sv-and-fusion/
- https://pecan.stjude.cloud/about
References¶
Activities¶
Initial Data Summary¶
# Summary of filetypes:
find . -type f | perl -ne 'print $1 if m/\.([^.\/]+)$/' | sort | uniq -c | tee AML-filetypes-summary.lst
# List all filenames:
find . -type f -exec basename {} \; | sort | tee AML-files-sorted.lst
Dataset Sizing¶
- https://groups.google.com/a/globus.org/g/archive-user-discuss/c/SWmHaxEB_v0
# 1. List data on an endpoint globus ls -Fjson ddb59aef-6d04-11e5-ba46-22000b92c6ec # 2. Filter the results to files only, no directories. # The Transfer API supports more advanced filtering, but the CLI's # `--filter` option only does filtering by name. So we'll use # jmespath to do the filtering client-side. globus ls ddb59aef-6d04-11e5-ba46-22000b92c6ec --jmespath 'DATA[?type==`file`]' # 3. Collect file sizes and convert the jmespath output to "unix" format globus ls ddb59aef-6d04-11e5-ba46-22000b92c6ec --jmespath 'DATA[?type==`file`].size' --format unix # 4. Sum the values (add `--recursive` to get a recursive listing) globus ls ddb59aef-6d04-11e5-ba46-22000b92c6ec --jmespath 'DATA[?type==`file`].size' --format unix | tr '\t' '\n' | awk '{sum+=$1}END{print sum}'
Summarizing VCFs¶
# List samples
bcftools query -l file.bcf
# Number of samples
bcftools query -l file.bcf | wc -l
#List of positions
bcftools query -f '%POS\n' file.bcf
Tagging¶
VCFs will be tagged with the following annotations: - clinVar - Cosmic - exAC/genomAD? - HGMD? - refGene - icgc28
References¶
- https://ocg.cancer.gov/news-publications/e-newsletter-issue/issue-20#2602
- TARGET Project Website
- TARGET AML Project Website
- https://ocg.cancer.gov/data/data-access